Mitochondria and lifespan control
An intuitive concept in human experience is that babies are born young, largely independent of the age of their parents. The finding that this phenomenon, mother-daughter age asymmetry, occurs in single-celled organisms like budding yeast led to a model in which aging factors are asymmetrically inherited during cell division. This asymmetric inheritance results in continued aging in mother cells and rejuvenation of daughter cells.
Indeed, aging determinants including oxidatively damaged proteins and extrachromosomal rDNA circles are selectively retained in yeast mother cells, while rejuvenating determinants including higher-functioning vacuoles (lysosomes) as well as activated antioxidants are preferentially inherited by yeast daughter cell. Moreover, defects in the asymmetric inheritance of any of these determinants compromises mother-daughter age asymmetry and lifespan control.
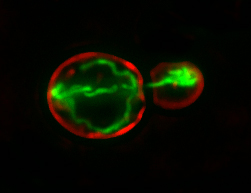
Using biosensors to monitor mitochondrial function in living yeast cells, we find that fitter mitochondria that are more reduced, have less reactive oxygen species, higher membrane potential and are more motile are preferentially inherited by yeast daughter cells.
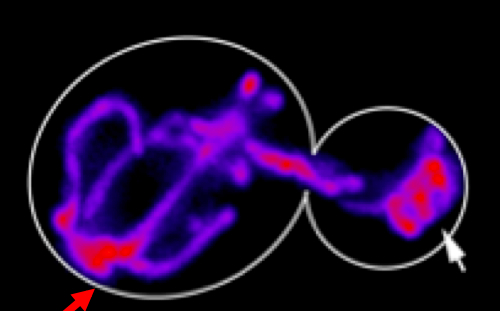
Equally important, we find that conditions that inhibit inheritance of fitter mitochondria by yeast daughter cells results in premature aging and loss of mother-daughter age asymmetry. Recent studies indicate that young and old mitochondria are segregated during cell division in human mammary stem-like cells and that this process affects cell fate. Specifically, daughter cells that inherit young mitochondria retain their stem cell properties, while cells that inherit old mitochondria differentiate. Thus, our studies on mitochondrial quality control during asymmetric inheritance in yeast may extend our understanding of similar processes in other eukaryotes.
Ongoing studies focus on the mechanisms for mitochondrial quality control. Specifically, we are studying the mechanism underlying the processes described below, how they affect lifespan and change as yeast cells age, and whether modulation of those processes can extend lifespan.
- Mitochondrial-cytoskeletal interactions and how they lead to preferential transport of fitter, more motile mitochondria from mother to daughter cells
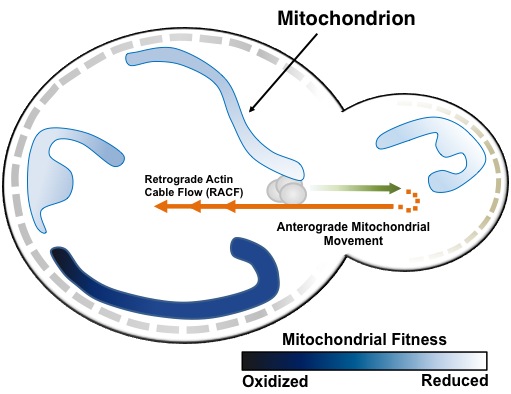
- Newly identified aging determinants that affect the actin cytoskeleton and mitochondria

Tethers that anchor and retain a population of higher functioning mitochondria in daughter cells

Role for lipid droplets and autophagy in relieving lipid imbalance and ER stress in response to lipid stress
The UPR (unfolded protein response) and one of its targets, ERAD (ER-associated protein degradation), are well-established mechanisms responding to ER stress (accumulation of unfolded proteins in the ER). Indeed, the UPR and ERAD play key roles in the immune response, brain development, learning and memory, and defects in these processes have been linked to diabetes, cancer and neurodegenerative diseases. We identified a novel pathway for ER quality control that relies on lipid droplets (LDs), organelles that contain a core of neutral lipids surrounded by a protein-containing phospholipid monolayer.
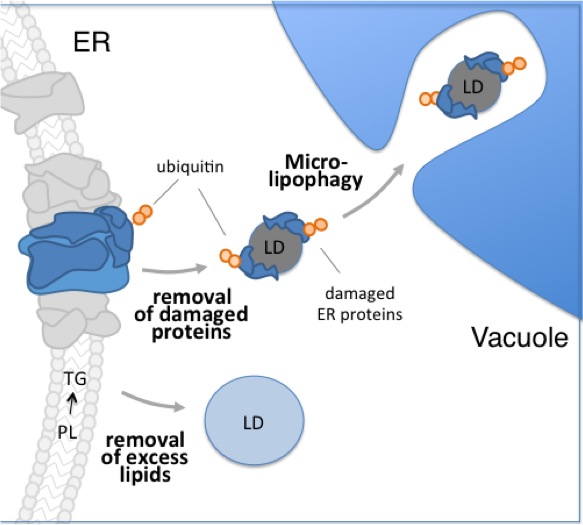
This pathway was initially identified in yeast undergoing acute lipid stress associated with defects in phosphatidylcholine (PC) biosynthesis. However, our preliminary evidence indicates that the pathway is also active in a mouse model for a congenital muscular dystrophy that is associate with defects in PC synthesis. In this pathway, ubiquitinated, presumably unfolded ER proteins are extracted from ER by LDs that form at and bud from the ER. LDs and their associated unfolded proteins are then degraded by autophagy. Interestingly, LD autophagy observed under these conditions does not occur by macroautophagy or require a core autophagy gene. Rather, LDs are degraded by a process that resembles microlipophagy, i.e. association of LDs with invaginations in the vacuolar membrane and direct uptake of LDs into the vacuole.
In all species studied, proteins associated with protein degradation and protein folding have been identified in LDs. Thus, it is possible that LD-mediated ER proteostasis is conserved in other eukaryotes. However, the mechanism underlying LD function in ER proteostasis is not well understood. Ongoing studies focus on the mechanism underlying microlipophagy in yeast and mouse disease models undergoing lipid stress, the consequence of defects in those processes, and whether interventions that promote ER quality control may serve as a foundation for treatment of lipid stress diseases.
